
ESKETAMINE JANSSEN 28 milligrammes, solution pour pulvérisation nasale, boîte de 1 flacon de 0,20 mL
Retiré du marché le : 30/03/2020
Dernière révision : 09/09/2019
Taux de TVA : 0%
Laboratoire exploitant : JANSSEN CILAG
Source :

Traitement des épisodes dépressifs caractérisés résistants n'ayant pas répondu à au moins deux antidépresseurs différents de deux classes différentes au cours de l'épisode dépressif actuel modéré à sévère, chez des adultes présentant une contre-indication à l'électroconvulsivothérapie (ECT) ou n'ayant pas accès à l'ECT ou étant résistant à l'ECT ou ayant refusé l'ECT. ESKETAMINE JANSSEN doit être coadministré avec un nouvel antidépresseur (AD) par voie orale.
·Hypersensibilité à la substance active, la kétamine, ou à l'un des excipients mentionnés à la rubriqueComposition.
·Patients pour qui une augmentation de la pression artérielle ou de la pression intracrânienne constitue un risque grave (voir rubrique Effets indésirables):
oPatients présentant une maladie vasculaire anévrismale connue (y compris des vaisseaux intracrâniens, thoraciques, ou de l'aorte abdominale, ou des artèrespériphériques)
oPatients présentant des antécédents connus d'hémorragieintracérébrale
Altérations des fonctions cognitives et motrices
Une somnolence, une sédation, des symptômes dissociatifs, des troubles de la perception, une sensation vertigineuse, des vertiges et une anxiété ont été rapportés avec ESKETAMINE JANSSEN au cours des essais cliniques (voir rubrique Effets indésirables). Ces effets peuvent altérer l'attention, le jugement, la pensée, la vitesse de réaction et les aptitudes motrices. Lors de chaque session de traitement, les patients doivent être placés sous la surveillance d'un professionnel de santé, qui évaluera si leur état est considéré comme cliniquement stable (voir rubrique Effets sur l'aptitude à conduire des véhicules et à utiliser des machines).
Suicide/idées suicidaires ou aggravation clinique
La dépression est associée à un risque accru d'idées suicidaires, d'auto-agressions et de suicide (comportement de type suicidaire). Ce risque persiste jusqu'à l'obtention d'une rémission significative, par conséquent les patients doivent être étroitement surveillés. L'expérience clinique montre que le risque suicidaire peut augmenter en tout début de rétablissement.
Les patients ayant des antécédents de comportements de type suicidaire ou ceux exprimant des idées suicidaires significatives avant de débuter le traitement sont connus pour être à risque plus élevé de survenue d'idées suicidaires ou de tentatives de suicide et ils doivent faire l'objet d'une surveillance étroite pendant le traitement.
Une surveillance étroite des patients, et en particulier de ceux à haut risque, doit accompagner le traitement, en particulier au début du traitement et lors des changements de dose. Les patients (et les aidants) doivent être avertis de la nécessité de surveiller la survenue d'une aggravation clinique, l'apparition d'idées ou comportements suicidaires et tout changement anormal du comportement et de prendre immédiatement un avis médical si ces symptômes survenaient.
Effet sur la pression artérielle
ESKETAMINE JANSSEN peut entrainer des augmentations transitoires de la pression artérielle systolique et/ou diastolique, avec un pic environ 40 minutes après l'administration du médicament et durant environ 1 à 2 heures (voir rubrique Effets indésirables). Les patients présentant une affection cardiovasculaire ou cérébrovasculaire doivent faire l'objet d'une évaluation attentive avant la prescription d'ESKETAMINE JANSSEN et le traitement ne doit être initié que si le bénéfice est supérieur au risque (voir rubrique Contre-indications). Des exemples d'affections nécessitant une attention particulière incluent :
·Hypertension artérielle instable ou malcontrôlée.
·Antécédents (dans les 6 semaines) d’événement cardiovasculaire, incluant l’infarctus du myocarde (IDM). Les patients présentant des antécédents d’IDM doivent être cliniquement stables et ne pas présenter de symptômes cardiaques avant l'administration du médicament.
·Antécédents (dans les 6mois) d'accident vasculaire cérébral ischémique ou d'accidentischémique transitoire.
·Cardiopathie valvulaire hémodynamiquement significative telle qu'une régurgitation mitrale, une sténose aortique ou une régurgitation aortique. Insuffisance cardiaque de Classe III ou IV selon la New York Heart Association (NYHA), quelle que soitl'étiologie.
L'administration
d’ESKETAMINE JANSSEN peut augmenter temporairement la pression
artérielle pendant une durée d’environ 1 à 2 heures. La pression
artérielle doit être mesurée avant l’administration d’ESKETAMINE
JANSSEN. Chez les patients dont la pression artérielle avant
l’administration est jugée élevée (à titre indicatif : >140/90 mmHg
pour les patients d’âge <65 ans et >150/90 mmHg pour les patients
d’âge ≥65 ans), une adaptation du mode de vie et/ou des traitements
pharmacologiques peuvent être envisagés pour réduire la pression
artérielle avant le démarrage du traitement par ESKETAMINE JANSSEN.
Chez les patients avec une pression artérielle élevée avant
l’administration d’ESKETAMINE JANSSEN, la décision de retarder le
traitement par ESKETAMINE JANSSEN doit être prise en tenant compte du
rapport bénéfice /risque pour chaque patient.
La pression artérielle doit être surveillée après l'administration de la dose. La pression artérielle doit être mesurée environ 40 minutes après l’administration de la dose d’ESKETAMINE JANSSEN et, par la suite selon les besoins cliniques jusqu’à ce que les valeurs baissent. Si la pression artérielle reste élevée sur une période de temps prolongée, il est nécessaire de solliciter rapidement l’assistance de personnel qualifié dans la prise en charge de la pression artérielle. Les patients présentant des symptômes de crise hypertensive doivent être immédiatement orientés vers un service d’urgence.
La pression artérielle doit être surveillée après l'administration de la dose. La pression artérielle doit être mesurée environ 40 minutes après l’administration de la dose d’ESKETAMINE JANSSEN et, par la suite selon les besoins cliniques jusqu’à ce que les valeurs baissent. Si la pression artérielle reste élevée sur une période de temps prolongée, il est nécessaire de solliciter rapidement l’assistance de personnel qualifié dans la prise en charge de la pression artérielle. Les patients présentant des symptômes de crise hypertensive doivent être immédiatement orientés vers un service d’urgence.
Abus, dépendance, sevrage
Les patients ayant des antécédents d'abus de substance ou de pharmacodépendance peuvent présenter un risque accru d'abus ou de mésusage d'ESKETAMINE JANSSEN.
Le risque d’abus ou de mésusage doit être évalué chez chaque patient avant la mise en place d’un traitement par ESKETAMINE JANSSEN et le développement de ces comportements ou états, y compris le comportement de recherche de drogues, doit être surveillé chez chaque patient pendant le traitement par ESKETAMINE JANSSEN..
Le risque d’abus, de mésusage ou d’usage détourné d’ESKETAMINE JANSSEN est minimisé par le fait que l’administration est réalisée sous la surveillance d’un professionnel de santé.
La kétamine, mélange racémique d’arkétamine et d’eskétamine, est un traitement pour lequel des abus ont été rapportés. ESKETAMINE JANSSEN peut être l’objet d’abus et d’usage détourné.
Des effets de dépendance et de tolérance ont été rapportés lors de l'utilisation prolongée de kétamine. Les personnes dépendantes à la kétamine ont décrit des symptômes de sevrage de type besoin irrépressible, anxiété, tremblements, sueurs et palpitations.
Des troubles cognitifs et des troubles de la mémoire ont été rapportés lors de consommation prolongée de kétamine ou d'abus de substances. Ces effets n'ont pas augmenté au cours du temps et étaient réversibles après l'arrêt du traitement par la kétamine. Lors des essais cliniques, l'effet de la pulvérisation nasale d'eskétamine sur le fonctionnement cognitif a été évalué au fil du temps et les performances cognitives sont restéesstables.
Effets sur la vessie
Des cas de cystite interstitielle ont été rapportés lors d’une utilisation quotidienne et à long terme de la kétamine à des doses élevées. Dans les études cliniques portant sur l’eskétamine en pulvérisation nasale, les symptômes de cystite, de douleur vésicale et de cystite interstitielle ont été évalués. Aucun cas de cystite interstitielle liée à l’eskétamine n’a été observé, dans aucune des études, impliquant un traitement d’une durée pouvant aller jusqu’à un an. Dans les études cliniques portant sur ESKETAMINE JANSSEN, un taux plus élevé de symptômes des voies urinaires inférieures (pollakiurie, dysurie, urgence mictionnelle, nycturie, et cystite) a été rapporté dans le groupe ESKETAMINE JANSSEN par rapport au groupe placebo..
Autres populations à risque
ESKETAMINE JANSSEN doit être utilisé avec prudence dans les situations suivantes. Les patients concernés doivent faire l'objet d'une évaluation attentive avant la prescription d'ESKETAMINE JANSSEN et le traitement ne doit être initié qu'après une évaluation favorable du rapport bénéfice/risque:
·Présence ou antécédent de psychose;
·Présence ou antécédent de manie ou de troubles bipolaires;
·Hyperthyroïdie non contrôlée;
·Insuffisance pulmonaire significative;
·Patients ayant une bradyarythmie ou tachyarythmie non contrôlée connue conduisant à une instabilité hémodynamique;
·Antécédents de lésion cérébrale, d'encéphalopathie hypertensive, de traitement intrathécal par shunts ventriculaires ou de toute autre affection associée à une augmentation de la pression intracrânienne.
Résumé du profil de sécurité
Les
effets indésirables les plus fréquemment rapportés chez les patients
présentant une dépression résistante traités par ESKETAMINE JANSSEN
étaient une dissociation (40%), une sensation vertigineuse (37%), des
nausées (27%), une sédation (25%), des céphalées (24%), des vertiges
(18%), une dysgueusie (17%), une hypoesthésie (17%), une augmentation
de la pression artérielle (13%), une anxiété (13%) et des vomissements
(10%).
Liste des effets indésirables
Les effets indésirables rapportés avec l'eskétamine sont listés dans le tableau ci-dessous. Au sein de chaque classe de systèmes d'organes, les effets indésirables sont listés par fréquence, en utilisant la convention suivante : très fréquent (≥ 1/10) ; fréquent (≥ 1/100 à < 1/10) ; peu fréquent (≥ 1/1 000 à<1/100); rare (≥1/10000à<1/1000); trèsrare (<1/10000); fréquence indéterminée (nepeutêtre estimée sur la base des données disponibles).
| Classe de systèmes d'organes | Effet indésirable | ||
| Fréquence | |||
| Très fréquent | Fréquent | Peu fréquent | |
| Affections psychiatriques | Dissociationa Anxiétéa | Humeur euphorique | |
| Affections du système nerveux | Dysgueusiea Sensation vertigineusea Sédationa Hypoesthésiea Céphaléea | Altération mentale Tremblements Léthargie Dysarthriea | |
| Affections de l'oreille et du labyrinthe | Vertigesa | ||
| Affections cardiaques | Tachycardiea | ||
| Affections respiratoires, thoraciques etmédiastinales | Inconfort nasala | ||
| Affections gastro-intestinales | Nausées Vomissements | Bouche sèche | Hypersécrétion salivaire |
| Affections de la peau et du tissu sous-cutané | Hyperhidrose | ||
| Affections du rein et des voies urinaires | Pollakiuriea | ||
| Troubles généraux et anomalies au site d'administration | Sensations anormales Sensation d'ébriété | ||
| Investigations | Pression artérielle augmentéea | ||
a Les termes suivants ont été combinés :
« Tachycardie » inclut : tachycardie sinusale ; tachycardie ; augmentation de la fréquence cardiaque ; extrasystole.
« Vertige » inclut : vertige ; vertige positionnel.
« Pression artérielle augmentée » inclut : pression artérielle augmentée, pression artérielle systolique augmentée ; pression artérielle diastolique augmentée ; hypertension ; cardiopathie hypertensive ; crise hypertensive.
« Sensation vertigineuse » inclut : sensation vertigineuse ; sensation vertigineuse orthostatique ; sensation vertigineuse procédurale ; sensation vertigineuse à l'effort.
« Dysarthrie » inclut : dysarthrie ; troubles de l'élocution ; élocution lente.
« Dysgueusie » inclut : dysgueusie ; hypogueusie.
« Hypoesthésie » inclut : hypoesthésie ; hypoesthésie buccale ; hypoesthésie dentaire ; hypoesthésie pharyngée ; hypoesthésie intranasale.
« Céphalée » inclut : céphalée ; céphalée sinusale.
« Léthargie » inclut : léthargie ; fatigue ; apathie.
« Sédation » inclut : sédation ; somnolence ; état de conscience altéré ; baisse du niveau de conscience ; hypersomnie ; stupeur.
« Tremblement » inclut : tremblement ; tremblement intentionnel.
«Anxiété»inclut: anxiété ; anxiété anticipatoire ; trouble anxieux ; trouble anxieux généralisé ; agitation ; peur ; nervosité ; tension ; crise de panique ; trouble panique ; réaction de panique ; sentiment de nervosité ; irritabilité ; tremblement psychogène.
« Dissociation » inclut : dissociation ; trouble de dépersonnalisation/déréalisation ; déréalisation ; trouble dissociatif ; flashback ; hallucination ; hallucination auditive ; hallucination visuelle ; illusion ; hallucination somatique ; hyperacousie ; acouphène ; diplopie ; vision trouble ; inconfort oculaire ; photophobie ; altération visuelle ; dysesthésie ; dysesthésie buccale ; paresthésie ; paresthésie buccale ; paresthésie pharyngée ; altération de la perception du temps ; rêverie ; perception délirante ; sensation de chaleur ; sensation de froid ; sensation de modification de la température corporelle.
« Pollakiurie » inclut : pollakiurie ; troubles mictionnels.
« Inconfort nasal » inclut : inconfort nasal ; croûtes nasales ; sécheresse nasale ; prurit nasal.
Description d'effets indésirables sélectionnés
Dissociation
La dissociation (40%) a été l’un des effets psychologiques les plus fréquents de l’eskétamine incluant une déréalisation (1,9%), une dépersonnalisation (1,7%), des illusions (1,5%) et une distorsion temporelle (1,2%). Ces effets indésirables ont été rapportés comme étant transitoires et auto-limitants et comme survenant le jour de l’administration. La dissociation a été rapportée comme d’intensité sévère à une incidence inférieure à 4 % dans les études. Les symptômes de dissociation ont généralement disparu dans un délai de 1,5 heures après l’administration et une tendance à la diminution de la sévérité a été observée avec le temps lors de traitements répétés.
Sédation/Somnolence
Les effets indésirables de type sédation (9,1%) et somnolence (18,0%) étaient le plus souvent de sévérité légère ou modérée, sont survenus le jour de l'administration et ont disparu spontanément le jour même. Les effets sédatifs disparaissent généralement dans un délai d'1,5 heures après l'administration.Lestauxdesomnolenceontétérelativementstablesdansletempslorsd'untraitement à long terme. Dans les cas de sédation, il n'a pas été observé de symptômes de détresse respiratoire et les paramètres hémodynamiques (incluant les signes vitaux et la saturation en oxygène) sont restés dans les limites de lanormale.
Modifications de la pression artérielle
Les augmentations moyennes de la pression artérielle systolique et de la pression artérielle diastolique (PAS et PAD) ajustées au placebo au fil du temps étaient d’environ 7 à 9 mmHg pour la PAS et d’environ 4 à 6 mmHg pour la PAD 40 minutes après l’administration, et de 2 à 5 mmHg pour la PAS et de 1 à 3 mmHg pour la PAD 1,5 heures après l’administration chez les patients recevant ESKETAMINE JANSSEN et des antidépresseurs oraux(voir rubrique Mises en garde et précautions d'emploi).Lafréquencedesélévations de la pression artérielle nettement anormales au cours des essais cliniques dans le traitement de la dépression résistante est présentée dans le Tableau2.
| Tableau 2: Elévations de la pression artérielle dans les études à court terme en double aveugle, randomisées et contrôlées comparant ESKETAMINE JANSSEN + antidépresseur (AD) par voie orale et placebo en pulvérisation nasale + antidépresseur(AD)parvoieoraledansletraitementdeladépressionrésistante | ||||
| Patients <65 ans | Patients ≥65 ans | |||
| ESKETAMINE JANSSEN + AD oral N=346 | Placebo + ADoral N=222 | ESKeTAMINE JANSSEN + AD oral N=72 | Placebo + ADoral N=65 | |
| Pression artérielle systolique | ||||
| ≥180 mmHg | 9 (3%) | --- | 2 (3%) | 1 (2%) |
| Augmentation ≥40 mmHg | 29 (8%) | 1 (0,5%) | 12 (17%) | 1 (2%) |
| Pression artérielle diastolique | ||||
| ≥110 mmHg | 13 (4%) | 1 (0,5%) | --- | --- |
| Augmentation ≥25 mmHg | 46 (13%) | 6 (3%) | 10 (14%) | 2 (3%) |
Sens de l'odorat
Le sens de l'odorat a été évalué au cours du temps ; aucune différence n'a été observée entre les patients traités par ESKETAMINE JANSSEN plus AD par voie orale et ceux traités par placebo en pulvérisation nasale plus AD par voie orale pendant la phase d'entretien en double aveugle de TRD3003.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté au moyen de la fiche correspondante (voir Annexe C) du protocole d’utilisation thérapeutique.
AVANT la mise en place du traitement : évaluer le risque d'abus ou de mésusage du patient.
SURVEIILANCE du traitement : la pression artérielle doit être mesurée avant l'administration, puis être réévaluée environ 40 minutes après l'administration et par la suite si nécessaire.INFORMER IMMEDIATEMENT le médecin en cas de :
- Comportements ou d'idées suicidaires ou de changement anormal du comportement.
- Douleur à la poitrine, essoufflement, maux de tête sévères et soudains, changement de la vision ou convulsions.
- Somnolence, évanouissement, vertiges, sensation de rotation, anxiété
ou sentiment de déconnexion de soi-même, de ses pensées, de ses sentiments,
de l'espace et du temps.
- Sang dans les urines ou douleur lors de l'évacuation de
l'urine.
EVITER le prise de boissons alcoolisées et de médicaments contenant de l'alcool.
NE PAS CONDUIRE ou UTILISER DES MACHINES avant le lendemain de la prise de ce médicament et un repos réparateur (somnolence, sédation, symptômes dissociatifs, troubles de la perception, sensation vertigineuse, vertiges et anxiété).
En
cas de prise concomitante un corticoïde nasal ou un décongestionnant nasal : ne
pas prendre ces médicaments dans l'heure précédant l'administration
d'eskétamine.
FEMME en AGE de procréer :
utiliser une méthode de contraception pendant le traitement et jusqu'à 6 semaines après la fin du traitement.
Grossesse
ESKETAMINE JANSSEN ne doit pas être utilisé pendant la grossesse. Il n'existe pas de données, ou seulement des données limitées, sur l'utilisation de l'eskétamine chez des femmes enceintes. Les études chez l'animal ont montré que la kétamine, mélange racémique d'arkétamine et d'eskétamine, induit une neurotoxicité chez les foetus en développement (voir rubrique Données de sécurité précliniques). Un risque similaire avec l'eskétamine ne peut pas être exclu.
ESKETAMINE JANSSEN n’est pas recommandé chez les femmes enceintes ou en âge de procréer n’utilisant pas de contraception. Si une femme débute une grossesse pendant le traitement par ESKETAMINE JANSSEN, le traitement doit être arrêté et la patiente doit être informée dès que possible du risque potentiel pour le foetus et des options cliniques/thérapeutiques.
Allaitement
ESKETAMINE JANSSEN ne doit pas être utilisé pendant l'allaitement. On ne sait pas si l'eskétamine estexcrétée dans le lait humain. Il n'existe aucune donnée disponible permettant d'évaluerles effetsde l'eskétamine sur la production de lait humain ou sur l'enfant allaité. Les données chez l'animal ont montré l'excrétion de la kétamine dans le lait de vache, en conséquence on peut s'attendre à ce que l'eskétamine soit également excrétée dans le lait humain. Il doit être conseillé aux patientes de ne pas utiliser ESKETAMINE JANSSEN pendant l'allaitement ou d'arrêter l'allaitement si le traitement par ESKETAMINE JANSSEN estinitié,en tenant compte de l'importance du traitement pour la mère.
Fertilité
Les études menées chez l'animal ont montré que l'eskétamine n'a pas d'effet sur la fertilité et la fonction de reproduction.
L'utilisation concomitante d'ESKETAMINE JANSSEN et de dépresseurs du système nerveux central (tels que benzodiazépines, opioïdes, alcool) peut augmenter la sédation. Surveiller attentivement la sédation lors de l'utilisation concomitante d'ESKETAMINE JANSSEN et de dépresseurs du système nerveux central.
L'utilisation concomitante d'ESKETAMINE JANSSEN avec des psychostimulants (tels que amphétamines, méthylphénidate, modafinil, armodafinil) peut augmenter la pression artérielle. Surveiller attentivement la pression artérielle lors de l'utilisation concomitante d'ESKETAMINE JANSSEN et depsychostimulants.
L'utilisation concomitante d'ESKETAMINE JANSSEN avec les inhibiteurs de la monoamine oxydase (IMAO) (tels que tranylcypromine, sélégiline, phénelzine) peut augmenter la pression artérielle. Surveiller attentivement la pression artérielle lors de l'utilisation concomitante d'ESKETAMINE JANSSEN etd'IMAO.
ESKETAMINE JANSSEN est destiné à être auto-administré par le patient sous la surveillance directe d'un professionnel de santé.
Une session de traitement consiste en une administration par voie nasale d'ESKETAMINE JANSSEN et une observation post-administration jusqu'à ce que le patient soit jugé cliniquement stable,en milieu hospitalier.
Évaluation de la pression artérielle avant et après traitement
La pression artérielle doit etre mesurée avant l'administration d'ESKETAMINE JANSSEN.
Si la pression artérielle initiale est élevée (par exemple > 140 mmHg systolique, > 90 mmHg diastolique),il faut prendre en compte les risques d'augmentation à court terme de la pression artérielle et le bénéfice du traitement par ESKETAMINE JANSSEN chez les patients présentant une dépression résistante (voir rubrique Mises en garde spéciales et précautions d'emploi). Ne pas administrer ESKETAMINE JANSSEN si une augmentation de la pression artérielle ou de la pression intracrânienne représente un risque grave (voir rubrique Contre-indications).
La pression artérielle doit être réévaluée environ 40 minutes après l'administration d'ESKETAMINEJANSSEN et par la suite sinécessaire.
Si la pression artérielle diminue et que le patient semble cliniquement stable, le patient peut quitter le milieu hospitalier à la fin de la période d'observation post-administration;sinon,la surveillance doit être poursuivie (voir rubriqueMises en garde et précautions d'emploi).
Recommandations concernant la prise d'aliments et de boissons avant l'administration du traitement
Etant donné que certains patients peuvent avoir des nausées et des vomissements après l’administration d’ESKETAMINE JANSSEN, il doit être conseillé aux patients de ne pas manger pendant au moins 2 heures avant l'administration et de ne pas boire de liquides pendant au moins 30 minutes avant l’administration (voir rubriqueEffets indésirables).
Corticoïdes par voie nasale ou décongestionnants par voie nasale
Il doit être conseillé aux patients ayant besoin d'utiliser un corticoïde nasal ou un décongestionnant nasal le jour d'une administration de ne pas prendre ces médicaments dans l'heure précédant l'administration d'ESKETAMINE JANSSEN.
Posologie
Les
recommandations posologiques d’ESKETAMINE JANSSEN sont fournies dans le
Tableau 1. Il est recommandé de maintenir la dose reçue par le patient
à la fin de la phase d’induction pendant la phase d’entretien. Les
adaptations posologiques doivent être faites en se basant sur
l’efficacité et la tolérance de la dose précédente. Pendant la phase
d’entretien, la posologie d’ESKETAMINE JANSSEN doit être individualisée
à la fréquence la plus basse pour maintenir la rémission/réponse.
| Tableau 1 : Posologie recommandée pour ESKETAMINE JANSSEN | |
| Phase d'induction | Phase d'entretien |
| Semaines 1 à 4*: | Semaines 5 à 8 : |
| Dose initiale au Jour 1 : < 65 ans : 56 mg | 56 mg ou 84 mg une fois par semaine |
| ≥ 65 ans : 28 mg | À partir de la semaine 9 : |
| Doses suivantes : 56 mg ou 84 mg 2 fois /semaine | 56 mg ou 84 mg toutes les 2 semaines ou une fois par semaine |
| Les signes indiquant un bénéfice thérapeutique doivent être évalués à la fin de la phase d'induction, afin de déterminer la nécessité de poursuivre le traitement. | La nécessité de poursuivre le traitement doit être régulièrement réexaminée. |
* Il n'est pas recommandé d'administrer ESKETAMINE JANSSEN pendant deux jours consécutifs.
Après l'amélioration des symptômes dépressifs, il est recommandé de maintenir le traitement pendant au moins 6 mois.
Observation post-administration
Pendant et après l’administration d’ESKETAMINE JANSSEN au cours de chaque session de traitement, les patients doivent être surveillés concernant les effets sédatifs et dissociatifs jusqu’à ce que l'état du patient soit jugé cliniquement stable (voir rubriqueMises en garde et précautions d'emploi).
Session(s) de traitement manquée(s)
Si le patient a manqué une ou deux sessions de traitement, la prochaine session doit être programmée à la date à laquelle la session de traitement suivante était programmée sur la base de la fréquence de traitement actuelle. Si le patient a manqué plus de 2 sessions de traitement, il peut être approprié sur le plan clinique d’adapter la dose ou la fréquence d'administration d’ESKETAMINE JANSSEN.
Populations particulières
Patients âgés (65 ans et plus)
Chez les patients âgés, la dose initiale d'ESKETAMINE JANSSEN est de 28 mg (Jour 1, dose initiale, voir Tableau 1 ci-dessus). Les doses suivantes doivent être augmentées par paliers de 28 mg jusqu'à 56 mg ou 84 mg, en fonction de l'efficacité et de la tolérance.
Insuffisance hépatique
Aucune adaptation posologique n'est nécessaire chez les patients présentant une insuffisance hépatique légère (classe A de Child Pugh) ou modérée (classe B de Child Pugh).
ESKETAMINE JANSSEN n'a pas été étudié chez des patients présentant une insuffisance hépatique sévère (classe C de Child Pugh). L'utilisation dans cette population n'est pas recommandée (voir rubrique Propriétés pharmacocinétiques).
Population pédiatrique
La sécurité et l’efficacité d’ESKETAMINE JANSSEN chez les patients pédiatriques âgés de 17 ans et moins n’ont pas été établies. Aucune donnée n’est disponible. L’utilisation d’ESKETAMINE JANSSEN dans l’indication du traitement de la dépression résistante chez les enfants de moins de 7 ans n’est pas justifiée.
Mode d'administration
ESKETAMINE JANSSEN est destiné à une utilisation nasale uniquement. Le dispositif pour pulvérisation nasale est un dispositif à usage unique, qui délivre un total de 28 mg d’eskétamine en deux pulvérisations (une pulvérisation par narine). Pour éviter la perte de médicament, le dispositif ne doit pas être amorcé avant utilisation. Il est destiné à être administré par le patient sous la surveillance d'un professionnel de santé, en utilisant 1 dispositif (pour une dose de 28 mg), 2 dispositifs (pour une dose de 56 mg) ou 3 dispositifs (pour une dose de 84 mg) avec une pause de 5 minutes entre l’utilisation de chaque dispositif.
Durée de conservation :
A conserver à une température ne dépassant pas 30°c
Ne pas mettre au réfrigérateur
2 ans
Précautions particulières de conservationA conserver à une température ne dépassant pas 30°c
Ne pas mettre au réfrigérateur
Sans objet.
Le risque de surdosage en ESKETAMINE JANSSEN par le patient est minimisé par la conception du produit et par le fait que l'administration est réalisée sous la surveillance d'un professionnel de santé (voir rubrique Posologie et mode d'administration).
Symptômes
La dose maximale d'eskétamine en pulvérisation nasale en une seule administration testée chez des volontaires sains était de 112 mg et n'a été associée à aucun signe de toxicité ni résultats cliniques défavorables. Toutefois, en comparaison à l'intervalle de doses recommandé, la dose de 112 mg d'eskétamine en pulvérisation nasale a été associée à des taux d'effets indésirables plus élevés, incluant : sensation vertigineuse, hyperhidrose, somnolence, hypoesthésie, sensations anormales, nausées et vomissements.
Des symptômes mettant en jeu le pronostic vital sont envisageables compte tenu de l'expérience avec la kétamine utilisée à une dose 25 fois supérieure à la dose utilisée habituellement en anesthésie. Les symptômes cliniques décrits sont des convulsions, arythmies cardiaques et arrêt respiratoire. L'administration par voie nasale d'une dose suprathérapeutique comparable d'eskétamine est peu probable.
Prise en charge
Il n'existe pas d'antidote spécifique pour le surdosage par l'eskétamine. En cas de surdosage, il faut envisager la possibilité d'une implication de plusieurs médicaments. La prise en charge du surdosage par ESKETAMINE JANSSEN doit consister en un traitement des symptômes cliniques et une surveillance appropriée. Une surveillance et un suivi rapprochés doivent être maintenus jusqu'à la guérison du patient.
Classe pharmacothérapeutique : Psychoanaleptiques ; autres antidépresseurs, code ATC : N06AX27.
Mécanisme d'action
L'eskétamine est l'énantiomère S de la kétamine racémique. Il s'agit d'un antagoniste non sélectif et non compétitif des récepteurs de la N-méthyl-D-aspartate (NMDA), récepteurs ionotropiques au glutamate. En tant qu'antagoniste des récepteurs N-méthyl-D-aspartate (NMDA), l'eskétamine produit une augmentation transitoire de la libération de glutamate, conduisant à des augmentations de la stimulation des récepteurs à acide α-amino-3-hydroxy-5-méthyl-4-isoxazolepropionique (AMPAR), entraînant à son tour des augmentations de la signalisation neurotrophique qui contribuent à la restauration de la fonction synaptique dans ces régions du cerveau impliquées dans la régulation de l'humeur et du comportement émotionnel. La restauration de la neurotransmission dopaminergique dans les régions du cerveau impliquées dans la récompense et la motivation et la diminution de la stimulation des régions du cerveau impliquées dans l'anhédonie, peuvent contribuer à la rapidité d'action.
Effets pharmacodynamiques
Risque d'abus
Dans une étude évaluant le risque d’abus des consommateurs de diverses drogues à des fins récréatives (n=41), les doses uniques d’eskétamine en pulvérisation nasale (84 mg et 112 mg) et de kétamine par voie intraveineuse (0,5 mg/kg perfusé sur 40 minutes) comme contrôle positif ont présenté des scores significativement plus élevés que le placebo dans les évaluations individuelles « d’appréciation de substances » et sur les autres perceptions subjectives du médicament.
Efficacité et sécurité clinique
L’efficacité et la sécurité de la pulvérisation nasale d’ESKETAMINE JANSSEN ont été évaluées dans cinq études cliniques de Phase 3 chez des patients adultes (âgés de 18 à 86 ans) présentant une dépression résistante selon les critères du DSM-5 définissant un épisode dépressif caractérisé et non répondeurs à au moins deux traitements antidépresseurs (AD) oraux administrés à une dose et pendant une durée adéquates, dans l’épisode dépressif caractérisé en cours. 1 833 patients adultes ont été recrutés, parmi lesquels 1 601 ont été exposés à ESKETAMINE JANSSEN.
Dépression résistante - Études à court terme
ESKETAMINE
JANSSEN a été évalué dans trois études de Phase 3 à court terme (4
semaines), randomisées, en double aveugle, contrôlées versus traitement
actif, chez des patients présentant une dépression résistante. Les
études TRANSFORM-1 (TRD3001) et TRANSFORM-2 (TRD3002) ont été conduites
chez des adultes (18 à < 65 ans) et l’étude TRANSFORM-3 (TRD3005) a
été conduite chez des adultes âgés de ≥ 65 ans. Les patients des études
TRD3001 et TRD3002 ont débuté le traitement
par ESKETAMINE JANSSEN 56 mg plus un nouvel AD oral en prise
quotidienne ou un nouvel AD oral en prise quotidienne plus placebo en
pulvérisation nasale au Jour 1. Les doses d’ESKETAMINE JANSSEN ont été
maintenues à 56 mg ou ajustées à 84 mg ou en correspondance au placebo
en pulvérisation nasale administrés deux fois par semaine durant une
phase d’induction en double aveugle de 4 semaines. Les doses
d’ESKETAMINE JANSSEN de 56 mg ou 84 mg étaient fixes dans l'étude
TRD3001 et flexibles dans l'étude TRD3002. Dans l'étude TRD3005, les
patients (≥ 65 ans) ont débuté le traitement par ESKETAMINE JANSSEN 28
mg plus un nouvel AD oral en prise quotidienne ou un nouvel AD oral en
prise quotidienne plus placebo en pulvérisation nasale (Jour 1).
Les doses d’ESKETAMINE JANSSEN ont été ajustées à une dose de 56 mg ou
84 mg ou en correspondance au placebo en pulvérisation nasale
correspondant administré deux fois par semaine durant une phase
d’induction en double aveugle de 4 semaines. Dans les études de doses
flexibles, TRD3002 et TRD3005, l’augmentation de la dose d’ESKETAMINE
JANSSEN était basée sur le jugement clinique et la dose pouvait être
réduite en fonction de la tolérance. Un nouvel AD oral (IRSN :
duloxétine, venlafaxine à libération prolongée ; ISRS : escitalopram,
sertraline) était initié en ouvert au Jour 1 dans toutes les études. Le
choix du nouvel AD oral était effectué par l’investigateur sur la base
des antécédents thérapeutiques du patient. Dans toutes les études à
court-terme, le critère d’efficacité principal était la modification du
score total MADRS de l’inclusion au Jour 28.
Les caractéristiques démographiques et de la maladie des patients à l'inclusion dans les études TRD3002, TRD3001, et TRD 3005 sont présentées dans le Tableau 3.
| Tableau3: Caractéristiques démographiques à l'inclusion des études TRD3002, TRD3001, et TRD 3005 (ensemble d'analyseintégral) | |||
| Etude TRD3002 (N=223) | Etude TRD3001 (N=342) | Etude TRD3005 (N=137) | |
| Age, années | |||
| Médiane (Intervalle) | 47,0 (19; 64) | 47,0 (18; 64) | 69,0 (65; 86) |
| Sexe, n (%) | |||
| Homme | 85 (38,1%) | 101 (29,5%) | 52 (38,0%) |
| Femme | 138 (61,9%) | 241 (70,5%) | 85 (62,0%) |
| Origine ethnique, n (%) | |||
| Blanc | 208 (93,3%) | 262 (76,6%) | 130 (94,9%) |
| Noir ou Afro-Américain | 11 (4,9%) | 19 (5,6%) | -- |
| AD oraux antérieurs avec non-réponse (i.e., échec d'antidépresseurs) | |||
| Nombre d'antidépresseurs spécifiques, n (%) | |||
| 2 | 136 (61,0%) | 167 (48,8%) | 68 (49,6%) |
| 3 ou plus | 82 (36,8%) | 167 (48,8%) | 58 (42,3%) |
| AD oral nouvellement initié à la randomisation, n (%) | |||
| ISRN | 152 (68,2%) | 196 (57,3%) | 61 (44,5%) |
| ISRS | 71 (31,8%) | 146 (42,7%) | 76 (55,5%) |
| Sortidel'étude(quellequesoit la raison), n/N(%) | 30/227 (13,2%) | 31/346 (9,0%) | 16/138 (11,6%) |
Dans l'étude à dose flexible TRD3002, au Jour 28, 67% des patients randomisés dans le groupe ESKETAMINE JANSSEN prenaient 84 mg. Dans l’étude TRD3002, eskétamine plus un nouvel AD oral ont été associés à une supériorité statistique et cliniquement significative comparativement à un nouvel AD oral (ISRN : duloxétine, venlafaxine à libération prolongée ; ISRS : escitalopram, sertraline) plus placebo en pulvérisation nasale (Tableau 4).
Dans l'étude TRD3001, un effet du traitement cliniquement significatif sur la modification des scores totaux MADRS entre l'inclusion et la fin de la phase d'induction de 4 semaines a été observé en faveur d'ESKETAMINE JANSSEN plus un nouvel AD oral par comparaison à un nouvel AD oral (ISRN : duloxétine, venlafaxine à libération prolongée ; ISRS : escitalopram, sertraline) plus placebo en pulvérisation nasale (Tableau 4).
Dans l'étude TRD3005, au Jour 28, 64% des patients randomisés dans le groupe ESKETAMINE JANSSEN prenaient 84 mg, 25 % prenaient 56 mg et 10% 28mg. Dans l'étude TRD3005, un effet du traitement cliniquement significatif sur la modification des scores totaux MADRS entre l'inclusion et la fin de la phase d'induction de 4 semaines a été observé en faveur d'ESKETAMINE JANSSEN plus un nouvel AD oral par comparaison à un nouvel AD oral (IRSN : duloxetine, venlafaxine à libération prolongée ; ISRS : escitalopram, sertraline) plus placebo en pulvérisation nasale (Tableau 4).
| Tableau 4 : Résultats du critère principal d'efficacité pour la modification du score total MADRS dans les essais cliniques de 4 semaines (BOCF (Baseline Observation Carried Forward) ANCOVA) | |||||
| Modification | |||||
| N° de l'étude | Groupe de traitement§ | Nombre de patients | Score moyen à l'inclusion (ET) | de la MMC entre l'inclusion et la fin dela Semaine 4 | Différence de MMC (IC à 95 %)† |
| (ES) | |||||
| TRD3001 | ESKETAMINE JANSSEN 56 mg + AD oral | 115 | 37,4 (4,8) | -19,7 (1,3) | -4,3 (-7,8, -0,8)# |
| ESKETAMINE JANSSEN 84 mg + AD oral | 114 | 37,8 (5,6) | -18,4 (1,3) | -1,2 (-4,7, 2,3)# | |
| AD oral + placebo en pulvérisation nasale | 113 | 37,5 (6,2) | -15,2 (1,3) | ||
| TRD3002 | ESKETAMINE JANSSEN (56 mg ou 84 mg) + AD oral | 114 | 37,0 (5,7) | - 19,0 (1,3) | -3,5 (-6,7, -0,3) ‡ |
| AD oral + placebo en pulvérisation nasale | 109 | 37,3 (5,7) | -15,6 (1,4) | ||
| TRD3005 (≥65ans) | ESKETAMINE JANSSEN (28 mg, 56 mg ou 84 mg) + AD oral | 72 | 35,5 (5,9) | -10,1 (1,7) | -2,9 (-6,5 -0,6)# |
| AD oral + placebo en pulvérisation nasale | 65 | 34,8 (6,4) | -6,8 (1,7) |
ET = écart type ; ES = erreur standard ; MMC = moyenne des moindres carrés ; IC = intervalle de confiance ; AD = antidépresseur
§ eskétamine ou placebo administré par voie nasale ; AD oral = un nouvel AD (ISRN : duloxétine, venlafaxine à libération prolongée ; ISRS : escitalopram, sertraline)
† Différence (ESKETAMINE JANSSEN + AD oral moins AD oral + placebo en pulvérisation nasale) de la moyenne des moindres carrés par rapport à l'inclusion
‡ Groupe de traitement qui était supérieur de manière statistiquement significative à AD oral + placebo en pulvérisation nasale
#Estimationnonbiaiséedelamédiane(c'est-à-direcombinaisonpondéréedesMMCdeladifférenceparrapport à AD oral + placebo en pulvérisation nasale), et intervalle de confiance flexible à 95%
Évolution de la réponse au traitement dans le temps
Dans l'étude TRD3002, un effet d'ESKETAMINE JANSSEN sur la réduction des symptômes a été observé dès 24 heures post-administration et a augmenté au cours des semaines suivantes ; l'effet antidépresseur complet d'ESKETAMINE JANSSEN était observé au Jour 28. La Figure 1 [Analyse BOCF ANCOVA avec barres d'ES] représente l'évolution dans le temps de la réponse pour le critère principal d'efficacité (MADRS) dans l'étude TRD3002. Un effet de traitement similaire a été observé dans les études TRD3001 et TRD3005.
Figure 1 : Modification de la moyenne des moindres carrés par rapport à l'inclusion du score total MADRS en fonction du temps dans l'étude TRD3002* (Ensemble d'analyse intégral) - Analyse BOCF ANCOVA avec barres d'erreur standard
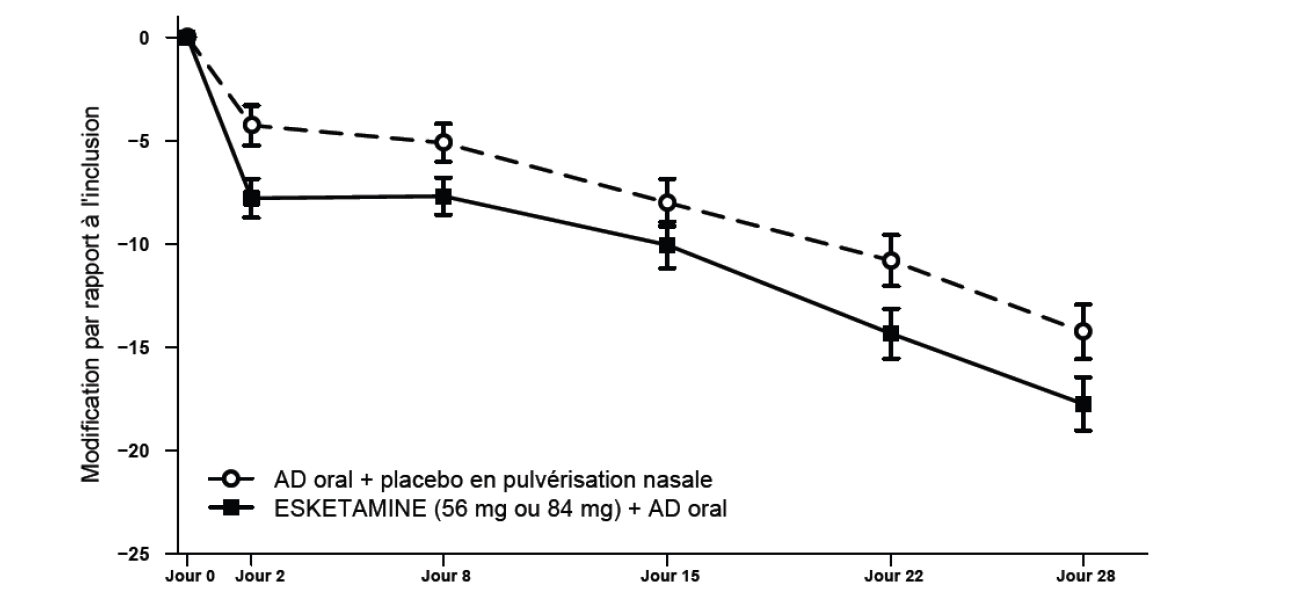
*Note : Dans cette étude à dose flexible, l'administration était individualisée sur la base de l'efficacité et de la tolérance. Chez un petit nombre de patients (< 10 %), la dose d'ESKETAMINE JANSSEN a été réduite de 84 mg à 56 mg et pratiquement tous les patients ont conservé la dose la plus faible pendant la durée de la phased'induction.
Taux de réponse et de rémission
La réponse était définie comme une réduction ≥ 50% du score total MADRS par rapport à l’inclusion dans la phase d’induction. Sur la base de la réduction du score total MADRS par rapport à l’inclusion, la proportion de patients dans les études TRD3001, TRD3002 et TRD3005 ayant obtenu une réponse à ESKETAMINE JANSSEN plus AD oral était supérieure à celle observée pour l’AD oral plus placebo en pulvérisation nasale tout au long de la phase d'induction en double aveugle de 4 semaines (Tableau5).
La rémission était définie comme un score total MADRS ≤ 12. Dans l’ensemble des trois études, une proportion supérieure de patients traités par ESKETAMINE JANSSEN plus AD oral était en rémission à la fin de la phase d'induction en double aveugle de 4 semaines comparativement à ceux recevant l’AD oral plus placebo en pulvérisation nasale (Tableau5).
| Tableau5: Tauxderéponseetderémissiondanslesessaiscliniquesde4semainessurlabasedesdonnées BOCF | |||||||
| N° de l'étude | Groupe de traitement§ | Nombre de patients (%) | |||||
| Taux de réponse† | Taux de rémission‡ | ||||||
| 24 heures | Semaine 1 | Semaine 2 | Semaine 3 | Semaine 4 | Semaine 4 | ||
| TRD3001 | ESKETAMINE JANSSEN 56 mg + AD oral | 20 (17,4 %) | 21 (18,3 %) | 29 (25,2 %) | 52 (45,2 %) | 61 (53,0 %) | 40 (34,8 %) |
| ESKETAMINE JANSSEN 84 mg + AD oral | 17 (14,9 %)# | 16 (14,0 %) | 25 (21,9 %) | 33 (28,9 %) | 52 (45,6 %) | 38 (33,3 %) | |
| AD oral + placebo en pulvérisation nasale | 8 (7,1 %) | 5 (4,4 %) | 15 (13,3 %) | 25 (22,1 %) | 42 (37,2 %) | 33 (29,2 %) | |
| TRD3002 | ESKETAMINE JANSSEN 56 mg | 18 (15,8 %) | 15 (13,2 %) | 29 (25,4 %) | 54 (47,4 %) | 70 (61,4 %) | 53 (46,5 %) |
| ou 84 mg + AD oral | |||||||
| AD oral + placebo en pulvérisation nasale | 11 (10,1 %) | 13 (11,9 %) | 23 (21,1 %) | 35 (32,1 %) | 52 (47,7 %) | 31 (28,4 %) | |
| TRD3005 (≥ 65ans) | ESKETAMINE JANSSEN 28 mg, 56 mg ou 84 mg + AD oral | ND | 4 (5,6 %) | 4 (5,6 %) | 9 (12,5 %) | 17 (23,6 %) | 11 (15,3 %) |
| AD oral + placebo en pulvérisation nasale | ND | 3 (4,6 %) | 8 (12,3 %) | 8 (12,3 %) | 8 (12,3 %) | 4 (6,2 %) |
AD = antidépresseur ; ND = non disponible
§ ESKETAMINE JANSSEN ou placebo administré par voie nasale ; AD oral = un nouvel AD (ISRN : duloxétine, venlafaxine à libération prolongée ; ISRS : escitalopram, sertraline)
† La réponse était définie comme une réduction ≥ 50% du score total MADRS par rapport à l'inclusion
‡ La rémission était définie comme un score total MADRS ≤12
# La première dose était ESKETAMINE JANSSEN 56 mg + AD oral
Dépression résistante- Études à long terme
Étude de prévention des rechutes
Le maintien de l’efficacité du traitement antidépresseur a été démontré dans un essai de prévention des rechutes. L'étude SUSTAIN-1 (TRD3003) était une étude de prévention des rechutes, à long terme, randomisée, en double aveugle, contrôlée versus traitement actif, avec groupes parallèles, multicentrique. Le critère principal d’évaluation, la prévention des rechutes dépressives, était mesuré par le délai avant rechute. Un total de 705 patients a été recruté : 437 recrutés directement ; 150 transférés de l'étude TRD3001 et 118 transférés de l'étude TRD3002. Les patients recrutés directement ont reçu ESKETAMINE JANSSEN (56 mg ou 84 mg deux fois par semaine) plus un AD oral dans une phase d’induction en ouvert de 4 semaines. A la fin de la phase d’induction en ouvert, 52% des patients étaient en rémission (score total MADRS ≤ 12) et 66% des patients étaient répondeurs (amélioration ≥ 50% du score total MADRS). Les patients répondeurs (455) ont continué de recevoir le traitement par ESKETAMINE JANSSEN plus AD oral dans une phase d'optimisation de 12 semaines. Après la phase d’induction, les patients recevaient ESKETAMINE JANSSEN chaque semaine pendant 4 semaines et à partir de la semaine 8, un algorithme (basé sur le MADRS) était utilisé pour déterminer la fréquence d’administration ; les patients en rémission (c’est-à-dire que le score total MADRS était ≤12) recevaient une dose toutes les 2 semaines ; toutefois, si le score total MADRS augmentait >12, alors la fréquence était augmentée à une administration hebdomadaire pour les 4 semaines suivantes ; avec pour objectif de maintenir les patients à la fréquence d’administration la plus basse pour le maintien de la rémission/réponse. Après la période de traitement de 16 semaines, les patients en rémission stable (n=176) ou réponse stable (n=121) ont été randomisés pour continuer le traitement par ESKETAMINE JANSSEN ou arrêter ESKETAMINE JANSSEN et passer au placebo en pulvérisation nasale. La rémission stable était définie comme un score total MADRS ≤12 pendant au moins 3 des 4 dernières semaines de la phase d’optimisation et une réponse stable était définie comme une réduction ≥50 % du score total MADRS par rapport à l’inclusion pendant les 2 dernières semaines de la phase d'optimisation, mais sans rémission stable.Rémission stable
Les patients en rémission stable ayant poursuivi le traitement par ESKETAMINE JANSSEN plus AD oral ont présenté un délai avant rechute des symptômes dépressifs plus long, de manière statistiquement significative, comparativement aux patients ayant reçu un nouvel AD oral (ISRN : duloxétine, venlafaxine à libération prolongée ; ISRS : escitalopram, sertraline) plus placebo en pulvérisation nasale (Figure 2). La rechute était définie comme un score total MADRS ≥22 pendant 2 semaines consécutives ou une hospitalisation pour une aggravation de la dépression ou tout autre événement cliniquement pertinent indicateur d’une rechute. Le délai médian avant rechute dans le groupe recevant un nouvel AD oral (ISRN : duloxétine, venlafaxine à libération prolongée ; ISRS : escitalopram, sertraline) plus placebo pulvérisation nasale était de 273 jours, tandis que le délai médian n'a pas pu être estimé pour ESKETAMINE JANSSEN plus AD oral, car ce groupe n’a jamais atteint un taux de rechute de 50 %..
Figure 2 : Délai avant rechute chez les patients en rémission stable dans l'étude TRD3003 (ensemble d'analyse intégral)
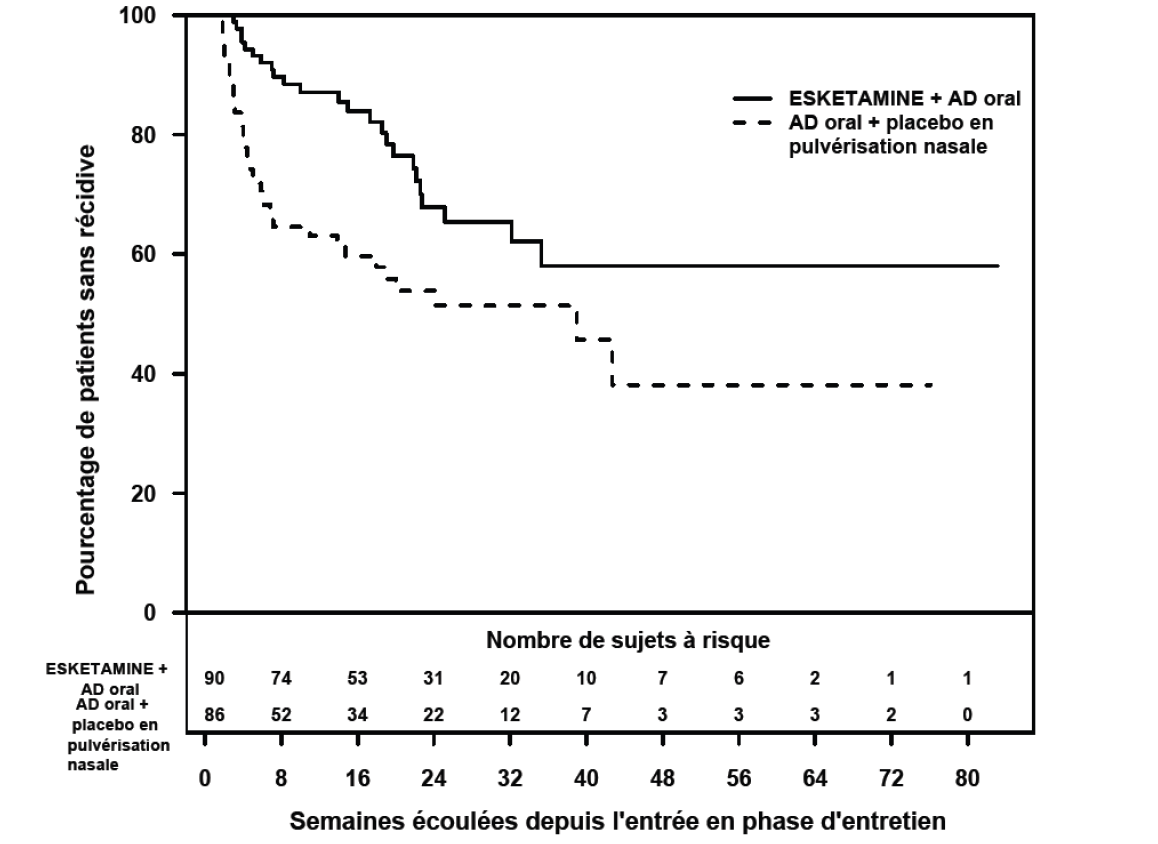
Pour les patients en rémission stable, le taux de rechute basé sur les estimations de Kaplan-Meier au cours des périodes de suivi de 12 et 24 semaines en double aveugle, était respectivement de 13% et 32% dans le groupe ESKETAMINE JANSSEN et de 37% et 46% dans le groupe placebo en pulvérisation nasale.
Réponse stable
Les résultats d’efficacité étaient également similaires pour les patients présentant une réponse stable ayant poursuivi le traitement par ESKeTAMINE JANSSEN plus AD oral ; les patients ont présenté un délai avant rechute des symptômes dépressifs plus long, de manière statistiquement significative, comparativement aux patients ayant reçu un nouvel AD oral (ISRN : duloxétine, venlafaxine à libération prolongée ; ISRS : escitalopram, sertraline) plus le placebo en pulvérisation nasale (Figure 3). Le délai médian avant rechute dans le groupe ayant reçu un nouvel AD oral (ISRN : duloxétine, venlafaxine à libération prolongée ; ISRS : escitalopram, sertraline) plus placebo en pulvérisation nasale (88 jours) était plus court que dans le groupe ESKETAMINE JANSSEN plus AD oral (635 jours).
Figure 3 : Délai avant rechute chez les patients présentant une réponse stable dans l'étude TRD3003 (ensemble d'analyseintégral)
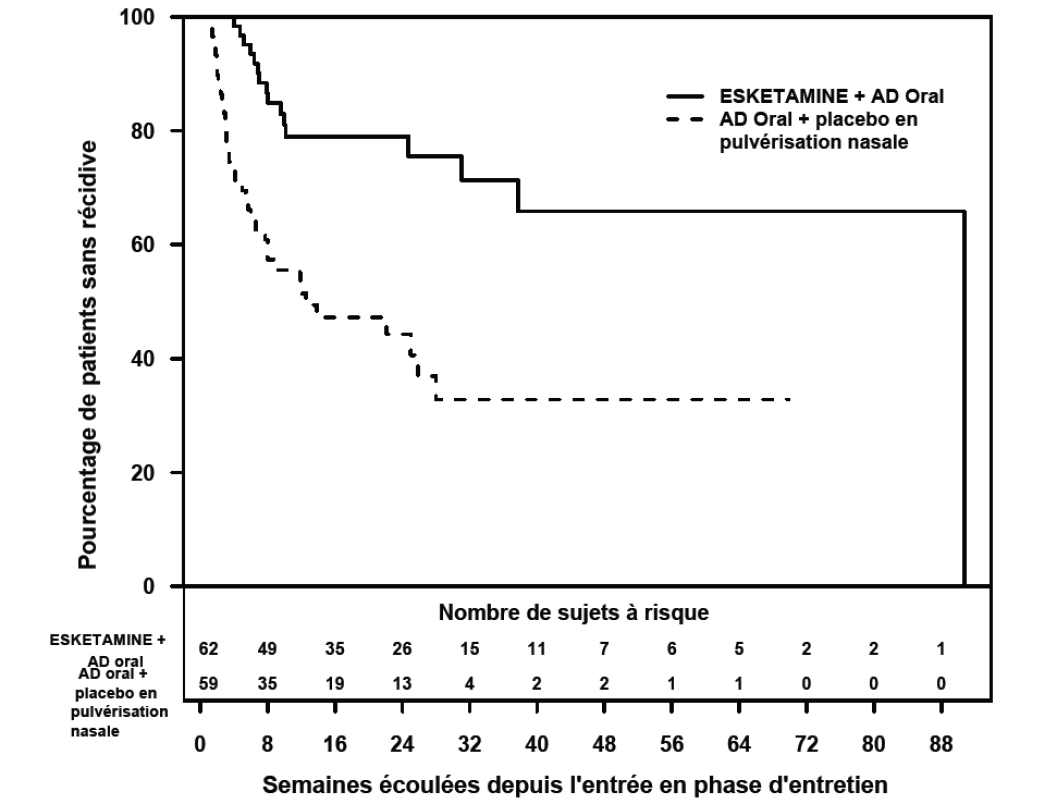
Pour les patients présentant une réponse stable, le taux de rechute basé sur les estimations de Kaplan- Meier au cours des périodes de suivi de 12 et 24 semaines en double aveugle, était respectivement de 21% et 21% dans le groupe ESKETAMINE JANSSEN et de 47% et 56% dans le groupe placebo en pulvérisation nasale.
Le
recrutement dans l'étude TRD3003 s’est échelonné sur environ 2 ans. La
phase d’entretien a été de durée variable et s’est poursuivie jusqu’à
ce que chaque patient présente une rechute des symptômes dépressifs ou
arrête pour toute autre raison, ou jusqu’à ce qu’il soit mis fin à
l'étude, le nombre d’événements de rechute requis ayant été atteint. Le
nombre d’expositions a été influencé par l'arrêt de l’étude à un nombre
prédéterminé de rechutes sur la base de l’analyse intermédiaire. Après
une période initiale de 16 semaines de traitement par ESKETAMINE
JANSSEN plus AD oral, la durée médiane d’exposition à ESKETAMINE
JANSSEN dans la phase d’entretien était de 4,2 mois (intervalle : 1
jour à 21,2 mois) chez les patients traités par ESKETAMINE JANSSEN
(rémission stable et réponse stable).
Dans cette étude, 31,6 % des patients ont reçu ESKETAMINE JANSSEN
pendant plus de 6 mois et 7,9 % des patients ont reçu ESKETAMINE
JANSSEN pendant plus d’1 an dans la phase d’entretien.
Fréquence d'administration
La fréquence d'administration utilisée la majorité du temps durant la phase d'entretien est présentée dans le Tableau 6. Parmi les patients randomisés pour recevoir ESKETAMINE JANSSEN, 60 % ont reçu la dose de 84 mg et 40 % ont reçu la dose de 56 mg.
| Tableau 6 : Fréquence d'administration utilisée la majorité du temps ; phase d'entretien (étude TRD3003) | ||||
| Rémission stable | Répondeurs stables | |||
| ESKETAMINE JANSSEN + AD oral (N=90) | AD oral + placebo en pulvérisation nasale (N=86) | ESKETAMINE JANSSEN + AD oral (N=62) | AD oral + placebo en pulvérisation nasale (N=59) | |
| Fréquence d'administration utilisée la majorité dutemps | ||||
| Chaque semaine | 21 (23,3 %) | 27 (31,4 %) | 34 (54,8 %) | 36 (61,0 %) |
| Tableau 6 : Fréquence d'administration utilisée la majorité du temps ; phase d'entretien (étude TRD3003) | ||||
| Rémission stable | Répondeurs stables | |||
| ESKETAMINE JANSSEN + AD oral (N=90) | AD oral + placebo en pulvérisation nasale (N=86) | ESKETAMINE JANSSEN + AD oral (N=62) | AD oral + placebo en pulvérisation nasale (N=59) | |
| Une semaine sur deux | 62 (68,9 %) | 48 (55,8 %) | 21 (33,9 %) | 19 (32,2 %) |
| Chaque semaine ou une semaine sur deux | 7 (7,8 %) | 11 (12,8 %) | 7 (11,3 %) | 4 (6,8 %) |
Absorption
La biodisponibilité absolue moyenne de 84 mg d'eskétamine administrés en pulvérisation nasale est d'environ 48 %.
L’eskétamine est rapidement absorbée par la muqueuse nasale après administration nasale et elle peut être mesurée dans le plasma dans les 7 minutes suivant une dose de 28 mg. Le délai nécessaire pour atteindre la concentration plasmatique maximale (tmax) est généralement de 20 à 40 minutes après la dernière pulvérisation nasale d’une session de traitement (voir rubrique Posologie et mode d'administration).
Les doses de 28 mg, 56 mg et 84 mg ont entrainé des augmentations dose-dépendantes de la concentration plasmatique maximale (Cmax) et de l'aire sous la courbe de la concentration plasmatique en fonction du temps (ASC∞) de l'eskétamine en pulvérisation nasale.
Leprofilpharmacocinétiquedel'eskétamineestsimilaireaprèsl'administrationd'unedoseuniqueetde doses répétées, sans accumulation dans le plasma lorsque l'eskétamine est administrée deux fois par semaine.
Distribution
Le volume de distribution moyen à l'équilibre de l’eskétamine administrée par voie intraveineuse est de 709 L.La proportion de la concentration totale d’eskétamine qui est liée à des protéines dans le plasma humain est en moyenne de 43 à 45 %. Le degré de liaison de l’eskétamine aux protéines plasmatiques ne dépend pas de la fonction hépatique ou rénale.
L’eskétamine n’est pas un substrat des transporteurs P-glycoprotéine (P-gp ; protéine de résistance multi-médicamenteuse 1), de la protéine de résistance du cancer du sein (BCRP), ou du transporteur d'anions organiques (OATP) 1B1, ou OATP1B3. L’eskétamine n’inhibe pas ces transporteurs ni les protéines d’extrusion de multiples médicaments et toxines (MATE1) et MATE2-K, ni le transporteur de cations organiques 2 (OCT2), OAT1, ou OAT3.
Biotransformation
L'eskétamine est largement métabolisée dans le foie. La principale voie métabolique de l'eskétamine dans les microsomes hépatiques humains est la N-déméthylation pour former la noreskétamine. Les principales enzymes du cytochrome P450 (CYP) responsables de la N-déméthylation de l'eskétamine sont le CYP2B6 et le CYP3A4. D'autres enzymes du CYP, incluant les CYP2C19 et CYP2C9, contribuent dans une mesure nettement plus faible. La noreskétamine est ensuite métabolisée par des voies dépendantes du CYP en d'autres métabolites, dont certains subissent une glucuronidation.
Élimination
La clairance moyenne de l’eskétamine administrée par voie intraveineuse était d’environ 89 L/heure. Une fois la Cmax atteinte après administration nasale, la baisse des concentrations d’eskétamine dans le plasma a été rapide pendant les quelques premières heures, puis plus progressive. La demi-vie terminale moyenne après administration sous forme de pulvérisation nasale était généralement comprise entre 7 et 12 heures..
Après administration
intraveineuse d’eskétamine marquée par un isotope radioactif, environ
78 % et 2 % de la radioactivité administrée étaient respectivement
retrouvés dans les urines et les fèces. Après administration orale
d’eskétamine marquée par un isotope radioactif, environ 86 % et 2 % de
la radioactivité administrée étaient respectivement retrouvés dans les
urines et les fèces. La radioactivité retrouvée consistait
essentiellement en des métabolites de l’eskétamine. Pour les voies
d'administration intraveineuse et orale, < 1% de la dose était
excrété dans l’urine sous forme de médicament inchangé.
Linéarité/non-linéarité
L’exposition à l’eskétamine augmente en fonction de la dose de 28 mg à 84 mg. L'augmentation des valeurs de Cmax et ASC étaient sensiblement proportionnelle à la dose entre 56 mg et 84 mg tandis que ce n'était pas le cas entre la dose de 28 mg et 56 ou 84 mg.
Interactions
Effets des autres médicaments sur l'eskétamine
Inducteurs des enzymes hépatiques
Le prétraitement par la rifampicine orale, un puissant inducteur de l'activité de nombreuses enzymes CYP hépatiques telles que CYP3A4 et CYP2B6 (600 mg par jour pendant 5 jours avant l’administration de l’eskétamine) a diminué les valeurs moyennes de Cmax et d’ASC∞ de l’eskétamine administrée par pulvérisation nasale de respectivement 17 % et 28 % environ.
Autres produits administrés par pulvérisation nasale
Le prétraitement de patients présentant des antécédents de rhinite allergique et préexposés au pollen de graminées par l’oxymétazoline administrée par pulvérisation nasale (2 pulvérisations de solution à0,05 % administrées 1 heure avant l’administration nasale de l’eskétamine) a eu des effets mineurs sur la pharmacocinétique de l’eskétamine.Le prétraitement de sujets sains par le furoate de mométasone administré par voie nasale (200 µg par jour pendant 2 semaines avec la dernière dose de furoate de mométasone administrée 1 heure avant l'administration nasale d’eskétamine) a eu des effets mineurs sur la pharmacocinétique de l’eskétamine.
Effet de l'eskétamine sur d'autres médicaments
L'administration
intranasale de 84 mg d’eskétamine deux fois par semaine pendant 2
semaines a réduit l’ASC∞ plasmatique moyenne du midazolam oral (dose
unique de 6 mg), un substrat du CYP3A4 hépatique, d’environ 16 %.
L'administration intranasale de 84 mg d’eskétamine deux fois par semaine pendant 2 semaines n'a pas eu d’incidence sur l’ASC plasmatique moyenne du bupropion oral (dose unique de 150 mg), un substrat du CYP2B6 hépatique.
Populations particulières
Personnes âgées (de 65 ans et plus)
La pharmacocinétique de l’eskétamine administrée en pulvérisation nasale a été comparée entre des sujets âgés, mais par ailleurs sains, et des adultes sains plus jeunes. Les valeurs moyennes de Cmax et d’ASC∞ de l’eskétamine produites par une dose de 28 mg étaient supérieures de respectivement 21 % et 18 % chez les sujets âgés (de 65 à 81 ans) par rapport aux sujets adultes plus jeunes (de 22 à 50 ans). Les valeurs moyennes de Cmax et d’ASC∞ de l’eskétamine produites par une dose de 84 mg étaient supérieures de 67 % et de 38 % chez les sujets âgés (de 75 à 85 ans) par rapport aux sujets adultes plus jeunes (de 24 à 54 ans). La demi-vie terminale de l’eskétamine était similaire chez les sujets adultes âgés et plus jeunes (voir rubriquePosologie et mode d'administration).
Insuffisance rénale
Par rapport aux patients présentant une fonction rénale normale (clairance de la créatinine [CLCR] 88 à 140 mL/min), la Cmax de l’eskétamine était supérieure de 20 à 26 % en moyenne chez les sujets présentant une insuffisance rénale légère (CLCR, 58 à 77 mL/min), modérée (CLCR, 30 à 47 mL/min) ou sévère (CLCR, 5 à 28 mL/min, pas sous dialyse) après l’administration d’une dose de 28 mg d'eskétamine en pulvérisation nasale. L’ASC∞ était supérieure de 13 à 36 % chez les sujets présentant une insuffisance rénale légère à sévère.
Il n'y a aucune expérience clinique avec l'eskétamine administrée en pulvérisation nasale chez des patients sous dialyse.
Insuffisance hépatique
La Cmax et l'ASC∞ de l'eskétamine produites par des doses de 28 mg étaient similaires entre les sujets présentant une insuffisance hépatique de classe Child-Pugh A (légère) et les sujets sains. LaCmax et l'ASC∞ de l'eskétamine étaient supérieures de respectivement 8 % et 103 % chez les sujets présentant une insuffisance hépatique de classe Child-Pugh B (modérée) par rapport aux sujets sains.
Il n'y a aucune expérience clinique avec l'eskétamine administrée en pulvérisation nasale chez des patients présentant une insuffisance hépatique de classe Child-Pugh C (sévère) (voir rubrique Posologie et mode d'administration).
Origine ethnique
La pharmacocinétique de l’eskétamine administrée en pulvérisation nasale a été comparée entre des sujets sains asiatiques et caucasiens. Les valeurs moyennes de Cmax et d’ASC∞ plasmatiques produites par une seule dose de 56 mg d’eskétamine étaient supérieures d’environ 14 % et 33 %, respectivement, chez les sujets chinois par rapport aux caucasiens. Les deux paramètres étaient supérieurs d’environ 40 % chez les sujets japonais par rapport aux caucasiens En moyenne, la Cmax de l’eskétamine était inférieure de 10 % et l’ASC∞ était supérieure de 17 % chez les sujets coréens comparativement aux caucasiens. La demi-vie terminale moyenne de l’eskétamine dans le plasma des sujets asiatiques était comprise entre 7,1 et 8,9 heures et elle était de 6,8 heures chez les caucasiens.
Sexe, poids corporel et insuffisance rénale
Aucune différence significative dans la pharmacocinétique d’ESKETAMINE JANSSEN en pulvérisation nasale n’a été observée due au sexe et au poids corporel total (> 39 à 170 kg) sur la base de l’analyse pharmacocinétique de population. Il n’existe aucune expérience clinique avec ESKETAMINE JANSSEN en pulvérisation nasale chez les patients en dialyse rénale.
Rhinite allergique
La pharmacocinétique d’une dose unique de 56 mg d’eskétamine administrée en pulvérisation nasale était similaire chez des sujets présentant une rhinite allergique exposés au pollen de graminées et chez des sujets sains.
ESKETAMINE JANSSEN a un effet important sur l’aptitude à conduire des véhicules et à utiliser des machines. Dans les études cliniques, une somnolence, une sédation, des symptômes dissociatifs, des troubles de la perception, une sensation vertigineuse, des vertiges et une anxiété ont été observés avec ESKETAMINE JANSSEN (voir rubrique Effets indésirables). Avant l'administration d'ESKETAMINE JANSSEN, les patients doivent être informés de ne pas entreprendre d'activités potentiellement dangereuses nécessitant une totale vigilance et une parfaite coordination motrice, comme la conduite d'un véhicule ou l'utilisation de machines, avant le lendemain et un repos réparateur (voir section Mises en garde et précautions d'emploi).
Les données non cliniques issues des études conventionnelles de toxicité après administration répétée, génotoxicité, neurotoxicité, toxicité de la fonction de reproduction et sur le potentiel cancérigène, n’ont pas révélé de risque particulier pour l’homme. Les études chez l’animal avec la kétamine ont montré des signes de neurotoxicité pour le développement. L’existence d’effets neurotoxiques potentiels de l’eskétamine sur les foetus en développement ne peut pas être exclue (voir rubriqueFertilité, grossesse et allaitement).
Génotoxicité
L’eskétamine ne s’est pas révélée mutagène avec ou sans activation métabolique dans le test d’Ames. Des effets génotoxiques avec l’eskétamine ont été observés dans un test de dépistage sur micronoyau in vitro en présence d’une activation métabolite. Toutefois, l’eskétamine administrée par voie intraveineuse ne possédait pas de propriétés génotoxiques dans un test in vivo du micronoyau de la moelle osseuse chez le rat et dans un essai Comet in vivo sur cellules hépatiques de rat.
Toxicité de la reproduction
Dans une étude de toxicité sur le
développement embryo-foetal avec la kétamine administrée par voie
nasale chez des rats, la descendance n’a pas été affectée de manière
défavorable en présence d’une toxicité maternelle à des doses allant
jusqu'à 150 mg/kg/jour. Chez les rats, à la dose de 150 mg/kg/jour de
kétamine, la marge de sécurité estimée pour l’eskétamine basée sur la
Cmax et l’ASC était 61 et 12 fois la dose maximale recommandée pour
l’eskétamine chez l’homme de 84 mg. Dans une étude de toxicité sur le
développement embryo-foetal avec la kétamine administrée par voie
intranasale chez des lapins, des malformations squelettiques ont été
notées à 30 et 100/50 mg/kg/jour en présence de toxicité maternelle. Le
lien avec le traitement par la kétamine ne peut être exclu. Chez les
lapins, l’exposition estimée à l’eskétamine à la dose sans effet de 10 mg/kg/jour était
inférieure à l’exposition maximale à l’eskétamine à la dose de 84 mg
chez l’homme.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Stupéfiant.
Prescription sur ordonnance sécurisée.
Prescription limitée à 28 jours.
Médicament réservé à l’usage hospitalier.
Médicament à prescription réservée aux spécialistes et services en psychiatrie.
Solution pour pulvérisation nasale.
Solution aqueuse transparente, incolore.
Le médicament est conditionné dans un flacon en verre de type I avec bouchon en caoutchouc chlorobutyle. Le flacon rempli et bouché est assemblé sous forme d'un dispositif pour pulvérisation nasale activé manuellement. Le dispositif permet de dispenser deux pulvérisations délivrant un volume total de 0,2 mL desolution.
ESKETAMINE JANSSEN est disponible en boites de 1 dispositif pour pulvérisation nasale.
Chaque dispositif est emballé individuellement dans un blister scellé.